What Are Design Controls in the Medical Device Industry?
Design controls are structured procedures implemented during medical device development to ensure safety, efficacy, and regulatory compliance. They are essential for meeting FDA, ISO 13485, and other global regulatory standards. These controls guide medical device manufacturers through every phase of the product lifecycle, from initial concept to production, ensuring that the device meets all necessary quality and safety standards.
Regulatory bodies, such as the FDA 21 CFR Part 820 and ISO 13485:2016, mandate design control processes as part of a company’s Quality Management System (QMS). Compliance with these requirements guarantees adherence to standards and builds a strong framework for systematic risk management and design documentation throughout the product development cycle.
Why Are Medical Device Design Controls Important?
Design controls are crucial for ensuring that medical devices are safe, effective, and meet regulatory requirements, particularly for Class II and III devices with greater risk profiles.
- Safety and Quality: Design controls provide a framework for identifying and mitigating risks early, ensuring that medical devices perform reliably.
- Regulatory Compliance: Adhering to standards like FDA 21 CFR Part 820 and ISO 13485:2016 ensures that medical devices meet both US and international quality and safety requirements. Non-compliance can result in recalls, fines, or other legal consequences.
- Consistent Documentation: Comprehensive design documentation ensures traceability and accountability throughout the process, fostering a strong quality system.
By integrating design controls, companies manage complex product development and ensure that each device passes rigorous quality checks before entering the market.
Design Control Phases
The design control process is divided into several key phases:
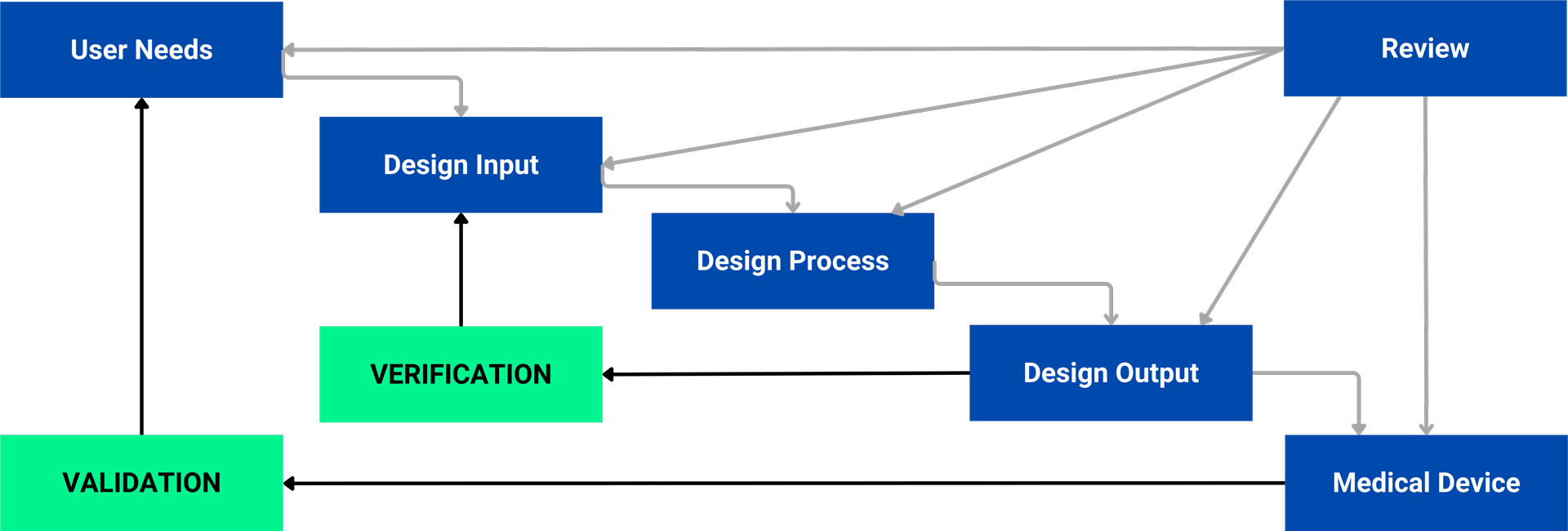
User Needs
This phase involves gathering detailed information about the device’s intended users and use cases. It ensures the device addresses the needs of physicians, patients, and caregivers while considering different environments (e.g., hospitals, clinics, or home settings).
Design Inputs
Here, broad user needs are transformed into specific, measurable design requirements. Design inputs must comply with FDA 21 CFR Part 820 and ISO 13485 standards. Key considerations include essential device functionalities, safety features, and risk mitigation strategies.
Design Outputs
Design outputs encompass the documentation necessary for manufacturing the medical device, including specifications, drawings, and the Bill of Materials (BOM). Ensuring that each output aligns with the corresponding design input is critical for maintaining traceability and compliance.
Design Reviews
Frequent design reviews help ensure that each phase meets the established requirements. Independent reviewers often objectively evaluate, identify misalignments, and suggest corrections as needed.
Design Verification and Validation
- Verification: Ensures the device meets the specified design inputs through rigorous testing, inspections, and measurements.
- Validation: Confirms that the device functions as intended in real-world conditions, typically involving clinical trials or usability testing to ensure safety and effectiveness.
Design Transfer
During design transfer, the focus shifts to ensuring that the device can be consistently manufactured while maintaining its intended quality. All necessary documentation, including the Device Master Record (DMR), must be complete and accurate. A final design review ensures the product is ready for full-scale production.
Key Regulations Affecting Design Controls
FDA 21 CFR Part 820
The 21 CFR Part 820 regulations outline the design control requirements for medical device manufacturers in the United States, emphasizing systematic development, risk management, and comprehensive documentation.
ISO 13485:2016
This international standard sets the framework for Quality Management Systems in medical devices, ensuring that products are safe, effective, and meet regulatory requirements. Certification under ISO 13485 demonstrates a company’s commitment to quality and compliance.

Choose Rook Quality Systems
At Rook Quality Systems, we specialize in helping medical device companies implement effective Design Controls in accordance with FDA 21 CFR Part 820 and ISO 13485. Our expertise ensures your products are developed efficiently, with robust verification, validation, and documentation processes, enabling a smooth path to market.
Schedule a consultation to learn more about how RookQS can help you with the design control process.