
This is Part 2 of a two-part series focusing on FDA’s Pre-Cert Working model.
4. Streamlined Review: It is expected that software products that are considered for Streamlined Review are from organizations that have gone through excellence Appraisal, and thus these have demonstrated excellence in developing, testing, maintaining, and improving software products. The streamlined review process is intended to identify what information would be reviewed, how modifications affect marketing authorization, and risk management established per device’s intended use and the SaMD’s clinical evaluation results (i.e. N41, SaMD Clinical Evaluation).
The process would work as follows:
a) Understand the product: While most of the elements are the same across all categories of the IMDRF, the depth of review changes with the risk of the product under review. Organization should include product-specific elements listed in Table.5 below as part of the packet for streamlined review application. The scope of a few elements is provided below, while the rest is defined on page 33 and 34.

-
Clinical Algorithm (including mechanism of action, design, development): This element provides adequate information on any clinical algorithms including mechanism of action.
-
Clinical Performance: The Clinical Performance of the device is product-specific and primarily reviewed during Streamlined Review for all risk levels. This would include the clinical data analysis and interpretation to demonstrate the adequate clinical performance of the device.
-
Cybersecurity: Cybersecurity is a Total Product Lifecycle activity. FDA evaluates the ability of the company to assess security, manage updates, perform post market security assessments of products, and proactively update products during the Excellence Appraisal and from Real-World Performance data. Streamlined Review would evaluate product-specific elements of cybersecurity to include how identified threats and mitigations impact the safety of the device.
-
Regulatory Pathway Specific Items: The Streamlined Review information would include, for example: a Substantial Equivalence (SE) comparison for 510(k) submissions to assess substantial equivalence to the identified predicate; an explanation of how special controls identified for a regulation are met; or a Risk/Benefit Analysis and proposed special controls for a De Novo application. Note that this is not a complete list of such items.
-
Validation (Performance Claims/Device Performance): Validation of the clinical algorithm is of primary importance and would be fully described and would include both protocols for testing and results demonstrating performance. For lower risk products, summary information for the validation information would be acceptable
b) Premarket review: A generic regulatory pathway in Figure.7 that depicts the generic process FDA would propose to follow in the Pre-Cert program, regardless of the regulatory pathway (i.e. 510(k) submission, De Novo Requests, and PMA applications). FDA would carry the information over from Review Determination into Streamlined Review and from the Pre-submission to reduce repetitive information. FDA would work interactively with the organization to understand the details of the software functions (i.e. SaMD product demo).
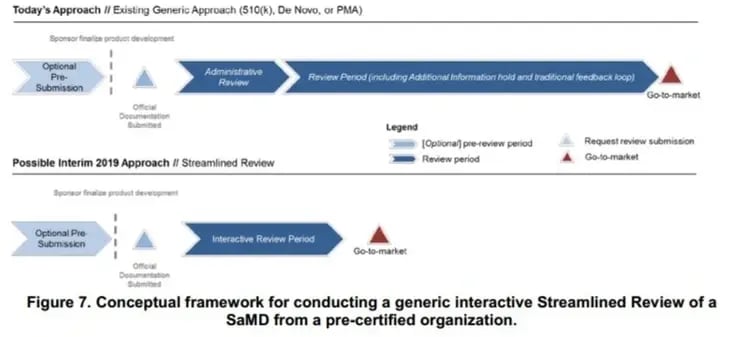
c) Marketing authorization: FDA would make a premarket decision, document a decision summary, retain the information and records on which the decision was based, and communicate the decision to the organization. FDA also indicates the occasions (Section 3.6) where an additional Excellence Appraisal is required, for instance, significant recurring patient and product issues.
For 2019 Testing, if FDA does not authorize the marketing of the product, the organization and FDA would complete an after-action review to determine gaps in the evidence supporting the submission and determine a plan for future submission. This will in turn help FDA review the basis of precertification to address any systemic issues within both the organization and the precertification program.
The objectives of Testing of Streamlined Review in 2019 include:
-
Determine the company’s and FDA’s responsibilities for interactive review as part of the Streamlined Review process.
-
Determine the most efficient method for conducting a product demo and determine how to capture that information in the review.
-
Develop a robust training program for FDA reviewers to make FDA Pre-Cert review as efficient and user-friendly as possible.
-
Continue to refine our approach to find a balance between internal resources, timely review, and reasonable assurance of safety and effectiveness.
5. Real-World Performance: Organizations must demonstrate the capability to collect and analyse post-launch real-world performance (RWP) data. Organization can reference Section 13 to identify RWP data elements for post-launch product monitoring. FDA intends to focus on its post-launch product monitoring efforts on trends and summary analytics, rather than on raw data. There are three major elements in this section:
-
Real-world health analytics: Analyses of real-world clinical output and outcomes related to the intended use of the SaMD product.
-
User experience analytics: Analyses of user experience outputs related to the real-world use of a SaMD product.
-
Product performance analytics: Analyses of outputs and outcomes demonstrating the real-world accuracy, reliability, and security of a SaMD product.
For 2019 Testing, FDA intends to review submitted RWPA collection plans and to work with each pilot participants to refine needed data elements. FDA would like to provide additional guidance to precertified organizations on appropriate types of analytics for verification. However, RWP Analytics framework is intended only for exploration and consideration for future Software Pre-Cert program and would not apply to SaMD products cleared by FDA prior to precertification of the organization. FDA expects that the 2019 testing and validation of the Pre-Cert framework would enable expanded uses of RWPA in future versions of the program.
The objectives of Testing of Real-World Performance in 2019 include:
-
Test processes and methodologies for data sharing and interpretation of analytics. Pilot Participants would provide FDA with access to RWPA, usage data, and software version information on a periodic basis (e.g., quarterly).
-
Refine the RWPA framework and provide additional clarity to industry on the attributes of RWP metrics that have high concordance with ongoing excellence.
-
Test the sensitivity of RWPA in detecting post market signals, as well as the alignment between RWPA signals and traditional post market reporting.
-
FDA expects that the 2019 testing and validation of the Pre-Cert framework would enable expanded uses of RWPA in future versions of the Program.
About the Author

Andrew Wu, Software Consultant at Rook Quality Systems.
“Working at the intersection of project and quality management, I believe better practice in software development and computer system validation can greatly benefit companies who are trying to put their novel medical technology in the hands of providers or patients”.
#FDA #SaMD #SDLC #Precertificationprogram #RealWorldPerformance #StreamlinedReview