
Summary of Key Points
-
Software development process require a variety of process planning activities that would address how you design, implement, and maintain software throughout its lifecycle.
-
Regulatory bodies require medical device software manufacturers to conform to a comprehensive list of guidelines, to make sure that no unacceptable risk introduced to patients with respect to safety and effectiveness.
-
Electronic QMS system can be a handy tool to achieve accurate documentation for regulatory audits and reviews.
Developing software is never an easy task, but maintaining software throughout its lifecycle could be a greater challenge, particularly in healthcare industry. Of all the 3140 medical devices recalled from 1992 to 1998, 7.7% of them are attributed to software. Of those software related recall, 79% were caused by software defects that were introduced when changes were made to the software after its initial production and distribution1. Software development process require a variety of process planning activities that would address how you design, implement, and maintain software throughout its lifecycle.
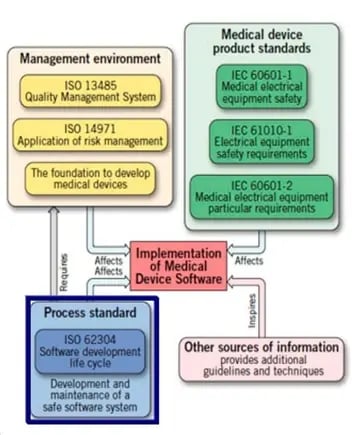
Regulatory bodies require medical device software manufacturers to conform to a comprehensive list of guidelines, to make sure that no unacceptable risk introduced to patients with respect to safety and effectiveness. All the guideline combined serve as a way to fully characterize, analyze, and mitigate unacceptable risks which software could expose patients to. Let’s simply the regulatory requirements to four pillars:
-
Management Environment: layout your QMS to run the project
-
Medical Device Product Standards: If the software is embedded in a medical device (i.e. Programmable Electrical Medical Equipment), the system level requirements and any collateral product standards can be found in IEC 60101-1 and 60101-2.
-
Process Standards: Use IEC 62304 as foundation while developing and maintaining a software system within a QMS.
-
Other Information: Usability requirement such as IEC 62366 and FDA guidance documents 1,2,3.
IEC 62304 defines software development lifecycle consists of the following stages:
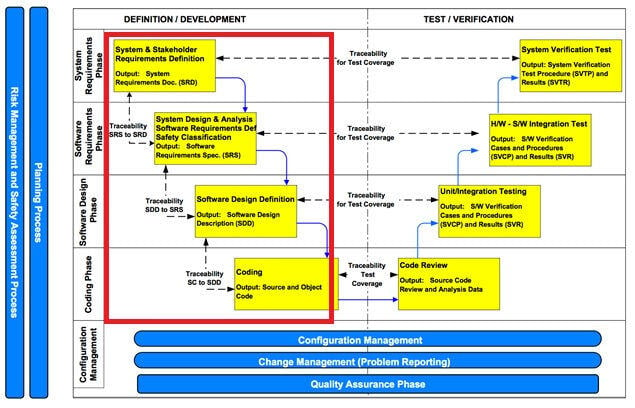
The key to develop a medical device software effectively is to start the risk management activity early, and applies recursively throughout software lifecycle. The typical risk management activity should be like this:
-
Identify item that could contribute to a hazardous situation
-
Identify causes of contribution (i.e. foreseeable events)
-
Designate software safety classification
-
Define risk control measures
-
Verify risk control measures
-
Document traceability
Soon as the system requirements are gathered and translated to software requirements, you will start designing and implementing the software architecture. The architecture is built upon building blocks, so called software items (i.e. back-end modules, front-end interface…etc.). Each software item should be mapped with a functional aspect of the system, and each function shall be classified differently per risk profile (please refer to IEC 62304 for detail). Developers can then use this ‘safety matrix’ to further refine system requirements and architecture accordingly.
If a potential software failure is identified, risk control measures such as a corrective/preventive/informative measure can be introduced to prevent possible failure of the system. Risk control measure can also lead to new requirements implemented in product hardware, software or documentation. For example, to prevent complete system failure, you can segregate software items that might collectively cause the failure. By segregating the items you have reduced the risk of failure by design. Developers should continuously monitor the system while being developed, and perform gap analysis numerous times to amend deficient processes and reduce risk.
Due to the complexity of medical device software, establishing an integrated change process across software lifecycle is absolutely critical. There will be many occasions to document software change control, version control, design review, and deployment activities. Innovative electronic QMS systems such as Greenlight.guru can be a handy tool to achieve accurate documentation for regulatory compliance and audits. After all, maintaining traceability from requirements through implementation and verification and validation activities is the backbone of good software development.
References:
1. General Principles of Software Validation
2. Guidance for the content of premarket submissions for software
3. Guidance for industry, FDA reviewers and compliance on Off-The-Shelf Software Use in Medical Devices.
4. Safety Classification from IEC 62304
About the Author

Andrew Wu, Software Consultant at Rook Quality Systems.
“Working at the intersection of project and quality management, I believe better practice in software development and computer system validation can greatly benefit companies who are trying to put their novel medical technology in the hands of providers or patients”.