
Link to the Policy Document: https://www.fda.gov/media/136290/download
As the COVID-19 situation is getting more serious all over the world, FDA issued an enforcement policy for non-invasive remote monitoring devices to combat this public health emergency.
SARS-CoV-2 (COVID-19) has demonstrated the capability to spread quickly. To respond effectively to the COVID-19 outbreak, FDA believes that the policy set forth in this guidance will help address this urgent public health concerns by helping to expand the availability and capability of remote patient monitoring devices. Modified use of these devices may increase access to important patient physiological data without the need for in-clinic visits and facilitate patient management by health care providers while reducing the need for in-office or in-hospital services during this time.
The policy applies to the following non-invasive remote monitoring devices:
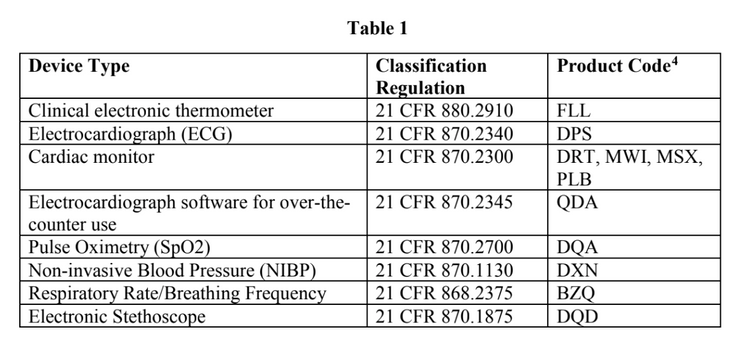
FDA does not intend to object to limited modifications to the indications, claims, functionality, or hardware or software FDA-cleared non-invasive remote monitoring devices that are used to support patient monitoring during the declared public health emergency without prior submission of a premarket notification[1].
A. Modifications to FDA-cleared Indications, Claims, or Functionality
FDA does not intend to object to modifications to the device indications, claims, or functionality where the modification does not create an undue risk in light of the public health emergency (e.g. intended for displaying, printing, and analyzing the physiological parameters measured by the device; intended for supporting or providing adjunctive recommendations; health care provider and/or patient can independently review the basis for any diagnostic or treatment recommendations).
Examples of circumstances where FDA currently believes a modification would create such an undue risk are:

FDA also layouts the ground work for labeling recommendations to help user better understand the device modifications on page 7 of the guidance.
B. Modifications to FDA-cleared Hardware or Software Intended to Increate Remote Monitoring Availability or Capability
FDA does not intend to object hardware or software architecture modifications that allow for increase remote monitoring capability to support additional claims or indications without prior submission of a premarket notification and where the modifications do not directly affect the physiological parameter measurement algorithms (e.g. BLE capability).
FDA recommends any such changes be designed, evaluated, and validated in accordance to FDA recognized standards on page 8 of the guidance.
C. Clinical Decision Support Software for Monitoring related to COVID-10 and Co-existing Conditions
Certain clinical decision support (CDS) software functions which are excluded from the definition of a device by section 520(o)(1)(E) of the FD&C Act are mentioned by this guidance. In the context of current public health emergency, FDA expect manufacturers to be aware of this section on non-device CDS in order to allow health care professionals or patients to gain access to software quickly that may be useful in connection with monitoring for patients with COVID-19 or co-existing conditions and providing clinical decision support.
This section excludes, from the definition of device, software functions that meet all of the following four criteria:

Examples of non-device functions under Section 520(o) provided in the guidance:

This policy is intended to remain in effect only for the duration of the public health emergency related to COVID-19 declared by the Department of Health and Human Services (HHS), including any renewals made by the Secretary in accordance with Section 319(a)(2) of the PHS Act.
Please reach out to RookQS team if you have any questions on this guidance or need help expanding the use of your current remote monitoring device.
[1] For further guidance on modifications that trigger the requirement that a manufacturer submit a new premarket notification (510(k)) to FDA, refer to “Deciding When to Submit a 510(k) for a Change to an Existing Device: Guidance for Industry and Food and Drug Administration Staff,” https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-when-submit-510k-change-existing-device.